
The power of machine learning models to learn the statistical representations underlying rich sequence and structural data provides new perspectives for protein structure prediction and design (41, 42, 44, 135) (Fig. 4D). Neural network models trained with evolutionary sequence data and structures from the PDB outperform traditional methods in structure prediction (41, 42, 135). Another approach using a deep convolutional neural network scoring function seeks to predict the probability distribution of amino acid types at each residue position conditioned on the local environment (102). Recent developments in binding site–generation methods aim to address these challenges.
AI Can Design Totally New Proteins From Scratch—It's Time to Talk Biosecurity - Singularity Hub
AI Can Design Totally New Proteins From Scratch—It's Time to Talk Biosecurity.
Posted: Mon, 29 Jan 2024 08:00:00 GMT [source]
Extended Data Fig. 8 Targeted unconditional and fold-conditioned protein binder design.
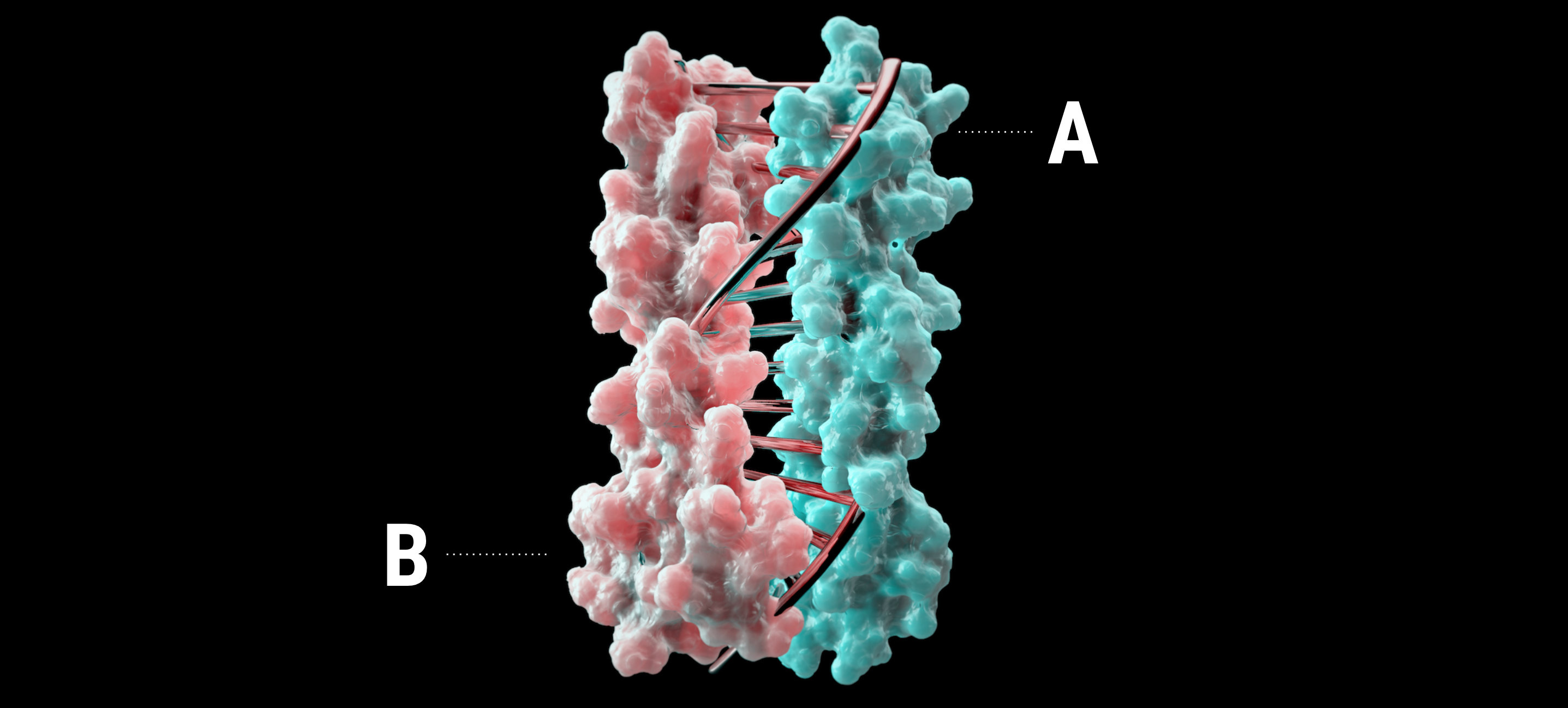
The designed proteins dimerized in the presence of the farnesyl pyrophosphate ligand and were able to transduce several modular downstream signals such as the enzyme activity, fluorescence, or luminescence. Latching orthogonal cage-key proteins is another recently designed protein switch system (15), consistent of a helical bundle and a helical peptide called key (Fig. 6D). The key peptide can displace a helix in the bundle and expose a signal on the displaced helix. The latching orthogonal cage-key proteins system was used to induce protein degradation and localization (15), target cells with precise combinations of surface antigens (23), and detect viral proteins (13). Protein design methods typically seek to find low-energy sequences for a given target structure, but this approach does not consider if there are alternative structures a sequence can adopt that have even lower free energies. One way to overcome this limitation is by directly calculating the fitness for a given structure in the protein sequence space.
Energy function
After generation of protein backbones, the second step in a typical de novo protein design protocol is selection of amino acid side-chain types and conformations to stabilize the backbone conformation and to adopt specific three-dimensional active site geometries optimized for function. Early de novo design studies used amino acids that favor specific secondary structure types (85) or binary polar/hydrophobic patterns (86) to define protein structures. Because side-chain conformations are clustered as rotamers (87, 88), the side-chain design can be formulated as a discrete optimization problem (89), that is, find a combination of rotamers that minimize the energy of a structure. The complexity of the problem grows exponentially with the increase of the number of residues. Small-scale side-chain design problems can be solved deterministically by the dead-end elimination algorithm (90), but many de novo protein side-chain optimization problems are too large to be solved deterministically. Instead, amino acid sequences and side-chain conformations are often optimized using Monte Carlo methods (91, 92), which do not guarantee to find the global minimum, but the solutions are often sufficiently accurate for applications.
A highly specific SpCas9 variant is identified by in vivo screening in yeast
A vaccine for COVID-19 that uses our protein design technology has been approved in the United Kingdom and South Korea. The vaccine is also the 12th COVID-19 vaccine in the world to be granted an Emergency Use Listing by the World Health Organization. Algorithm DevelopmentBiomolecular modeling and computational design are at the heart of everything we do.
A protein design algorithm must, thus, search all the conformations of each sequence, with respect to the target fold, and rank sequences according to the lowest-energy conformation of each one, as determined by the protein design energy function. Thus, a typical input to the protein design algorithm is the target fold, the sequence space, the structural flexibility, and the energy function, while the output is one or more sequences that are predicted to fold stably to the target structure. In addition to predicting protein structures, computational methods also allow scientists to simulate how proteins will interact with other molecules. This information is vital in refining protein designs to optimize their stability, binding affinity, or catalytic activity. Advanced software, like the Rosetta suite, empowers scientists to predict how a given amino acid sequence will fold into a three-dimensional structure.
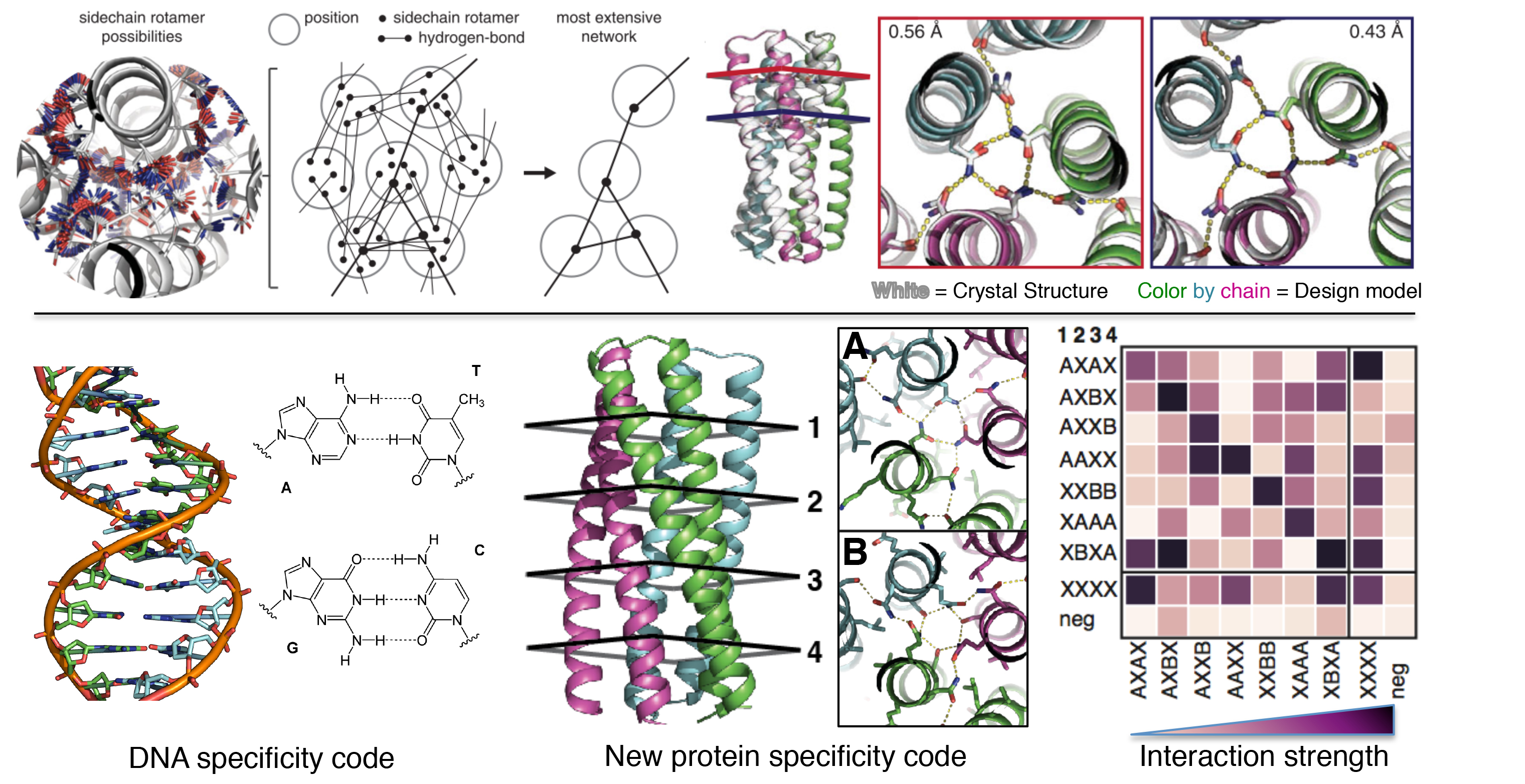
Encouraging results also have been achieved on orphan and de novo (designed) benchmarks with few homologous sequences. Furthermore, analysis conducted by the PDBench tool suggests that SPDesign performs well in subdivided structures. More interestingly, we found that SPDesign can well reconstruct the sequences of some proteins that have similar structures but different sequences.
We release OpenCRISPR-1 publicly to facilitate broad, ethical usage across research and commercial applications. Proteins perform functions by placing atoms with certain physicochemical properties at specific positions in the three-dimensional space. Initial work on the functional protein design directly borrowed from native functional site “motifs” (three-dimensional arrangements of functional groups in an existing active site) (136).
Improved cytosine base editors generated from TadA variants
We set out to generalize RFdiffusion to create symmetric oligomeric structures with any specified point group symmetry. For octahedral and icosahedral architectures, we explicitly model only the smallest subset of monomers required to generate the full assembly (for example, for icosahedra, the subunits at the five-, three- and twofold symmetry axes) to reduce the computational cost and memory footprint. First, when both physics- and knowledge-based terms are included in the force-field to generate and evaluate the designs, it is difficult to calibrate the relative strengths of the terms and determine the energy of the design in physical units. Thus, it would be preferable to rank designs using several metrics rather than using the same force-field that guided the design process. In addition, when using approaches that mix physics- and knowledge-based terms, it is difficult to ensure that all protein–protein and protein–water enthalpic contributions are properly accounted for and that the protein and solvent entropic contributions are included. Privett et al.23 addressed some of these issues by using all-atom MD simulations in addition to their standard design protocol (Figure 3 left), to assess iterative designs of a Kemp eliminase, resulting in a functional enzyme after three rounds.
For future in vivo applications, it would be useful to develop fluorescent proteins that are brighter and which mature faster than existing variants. In addition, proteins that emit at longer wavelengths would be beneficial to allow for deeper tissue penetration necessary for imaging in live multicellular organisms. For example, the mutations introduced to produce a monomeric VFP also decreased fluorescent brightness.62 Future design efforts will also focus on improving the fluorescent properties of novel proteins. This will be achieved through iterative rounds of design, solving crystal structures of the new protein, and then using these structures as a guide for further improvement. This approach has been used to improve the quantum yield of Cyan Fluorescent Protein (CFP) from 0.21 to most recently 0.93, the highest value to date for a monomeric protein.107 Improved selection procedures, as well as past experience, will expedite this process.
Applying design protocols on different problems and testing the methods systematically can be valuable for identifying and addressing limitations. Emerging machine learning methods provide opportunities and challenges in this relatively new subfield. Machine learning methods can not only synthesize existing data into statistical models that generate novel proteins but also iteratively integrate experimental data to guide the protein design (157). The best design strategies for many problems might be combinations of machine learning models and advances in existing design methods. There has been considerable recent progress in designing new proteins using deep-learning methods1,2,3,4,5,6,7,8,9.
The use of monoclonal bodies in the treatment of diseases has expanded dramatically as a result of the rising cases of chronic illnesses like cancer and other autoimmune diseases. Additionally, the region's top market competitors are concentrated on developing protein-engineered pharmaceuticals and vaccines, launching new products, and expanding the number of product approvals. These efforts are offering better treatment options, which is anticipated to increase market demand.
Automatic computational tools have enabled nonexperts to accurately design well-folded de novo proteins (71). Because proteins have highly diverse structures and functions, the difficulties of design problems also have great variations (Fig. 7, Tables 1 and and2).2). While robust protocols exist for designing helical bundles and small, idealized proteins with certain alpha-beta fold topologies (30, 58, 64), the success rates for other proteins such as beta barrels can be low (29, 31, 34). Addressing those challenging problems still requires significant amount of expertise, and sometimes trial and error.
One of these is a project with the Children's Hospital Zurich for the treatment of medulloblastomas, the most common malignant brain tumours in children. Moreover, the researchers have published the algorithm and its software so that researchers worldwide can now use them for their own projects. Because of the extended duration of action of insulin glargine, insulin can be released steadily and continuously over time. This was achieved by employing recombinant DNA technology to change the amino acid sequence of human insulin, which led to a prolonged therapeutic effect and a slower rate of absorption. A great label shows the world what you stand for, makes people remember your brand, and helps potential customers understand if your product is right for them.







No comments:
Post a Comment